When a generic drug hits the market, you expect it to work just like the brand-name version. But how do regulators know it really does? For simple pills, checking the peak concentration (Cmax) and total exposure (AUC) was enough. But for complex formulations-like extended-release opioids, abuse-deterrent pain meds, or mixed-mode drugs-those old metrics often miss the real story. That’s where partial AUC comes in.
Why Traditional Metrics Fall Short
The old way of proving bioequivalence relied on two numbers: how high the drug goes in your blood (Cmax) and how much total drug you’re exposed to over time (AUC). For immediate-release tablets, this worked fine. But for drugs designed to release slowly-like a 12-hour painkiller-you can’t just look at the total. Two products might have the same AUC and Cmax, but one releases the drug too fast at first, raising abuse risk, while the other releases it evenly. That difference matters clinically. The FDA and EMA realized this in the early 2010s. If a generic drug delivers the same total dose but gets absorbed too quickly in the first hour, it could be dangerous. Traditional AUC and Cmax couldn’t catch that.What Is Partial AUC?
Partial AUC, or pAUC, is a smarter way to measure drug exposure. Instead of looking at the whole curve from zero to infinity, you zoom in on a specific time window that matters most. For example, you might look only at the first 2 hours after dosing-the window where rapid absorption could lead to overdose or abuse. Or you might focus on the first 4 hours for a drug that needs to kick in fast for migraine relief. The idea is simple: focus on the part of the curve where differences between products could actually affect patient safety or effectiveness.The FDA started pushing pAUC after a 2013 draft guideline from the European Medicines Agency highlighted gaps in measuring extended-release formulations. By 2018, the FDA’s Center for Drug Evaluation and Research began standardizing how pAUC should be used across all drug types. Now, over 127 specific products have official guidance requiring pAUC analysis. That number is growing-FDA’s 2023 draft guidance added 41 more drugs.
How Is pAUC Calculated?
There’s no single formula. The time window for pAUC depends on the drug’s purpose. The FDA says the cutoff should be tied to a clinically relevant effect-like when the drug starts working or when abuse potential peaks. Common approaches include:- From time zero to the time when the reference product reaches its peak concentration (Tmax)
- From zero to 50% of Cmax, to capture the early absorption phase
- From zero to a fixed time (e.g., 1, 2, or 4 hours) based on pharmacodynamic data
Once the window is set, you calculate the area under the curve just for that interval. Then you compare the test product to the reference using the same 80-125% bioequivalence range. But here’s the twist: because pAUC focuses on a smaller part of the curve, the variability is often higher. That means you might need more people in your study. One Teva biostatistician reported increasing sample size from 36 to 50 subjects for an extended-release opioid generic-adding $350,000 to development costs. But it prevented a clinical failure.
Where Is pAUC Used Most?
pAUC isn’t needed for every drug. It’s reserved for products where absorption timing affects safety or effectiveness. The highest use is in:- Central nervous system drugs (68% of new submissions)-like ADHD meds and sleep aids where rapid onset can cause misuse
- Pain management (62%)-especially abuse-deterrent opioids where early high concentrations are dangerous
- Cardiovascular agents (45%)-where steady drug levels are critical to avoid arrhythmias or hypotension
In these cases, pAUC has caught problems traditional metrics missed. One 2021 AAPS case study showed a 22% difference in early exposure between a test and reference product. The total AUC was within limits, and Cmax was fine. But the early pAUC revealed the generic released too fast-potentially leading to overdose. The product was blocked before it reached patients.
Challenges and Controversies
Despite its value, pAUC isn’t easy to use. One big problem: inconsistency. The FDA’s product-specific guidances vary widely. Only 42% clearly explain how to pick the time window. That creates confusion for generic manufacturers. A Reddit post from a generic drug developer in 2023 complained: “There’s no standard. One drug says use 0-2 hours, another says 0-Tmax. How am I supposed to design a study?”Regulators have noticed. The International Consortium for Innovation and Quality in Pharmaceutical Development (IQ Consortium) found inconsistent pAUC rules across countries add 12-18 months to global drug approval timelines. And the statistics? They’re harder. Most bioequivalence specialists need 3-6 months of extra training to handle pAUC properly. A 2022 survey found 63% of companies needed outside statistical help for pAUC-compared to just 22% for traditional metrics.
There’s also the cost. Larger studies mean higher expenses. For small companies, outsourcing pAUC analysis to specialized CROs like Algorithme Pharma has become common. These firms now hold 18% of the complex generic bioequivalence market.
What’s Next for pAUC?
The trend is clear: pAUC is becoming standard for complex drugs. Evaluate Pharma predicts that by 2027, 55% of new generic approvals will require pAUC-up from 35% in 2022. The FDA is working on solutions. In January 2023, they launched a pilot program using machine learning to automatically determine optimal time windows based on reference product data. This could reduce subjectivity and make guidelines more consistent.For now, if you’re developing a generic version of an extended-release, abuse-deterrent, or mixed-mode drug, you can’t ignore pAUC. It’s not optional anymore. The science is solid. As Dr. Bingming Wang, FDA’s Director of Bioequivalence, said in 2022: “For some products, Cmax and AUC just aren’t enough.”
How to Get Started
If you’re new to pAUC, here’s how to begin:- Check the FDA’s product-specific guidances. Search for your drug in the 2,000+ published guidances-about 15% now mention pAUC.
- Identify the clinically relevant time window. Look at pharmacodynamic data: when does the drug start working? When does abuse risk peak?
- Use pilot data. Run small studies to find the reference product’s Tmax and variability.
- Plan for bigger studies. Expect to increase your sample size by 25-40% compared to traditional BE studies.
- Partner with experts. Hire a biostatistician experienced in pAUC or outsource to a CRO with proven methods.
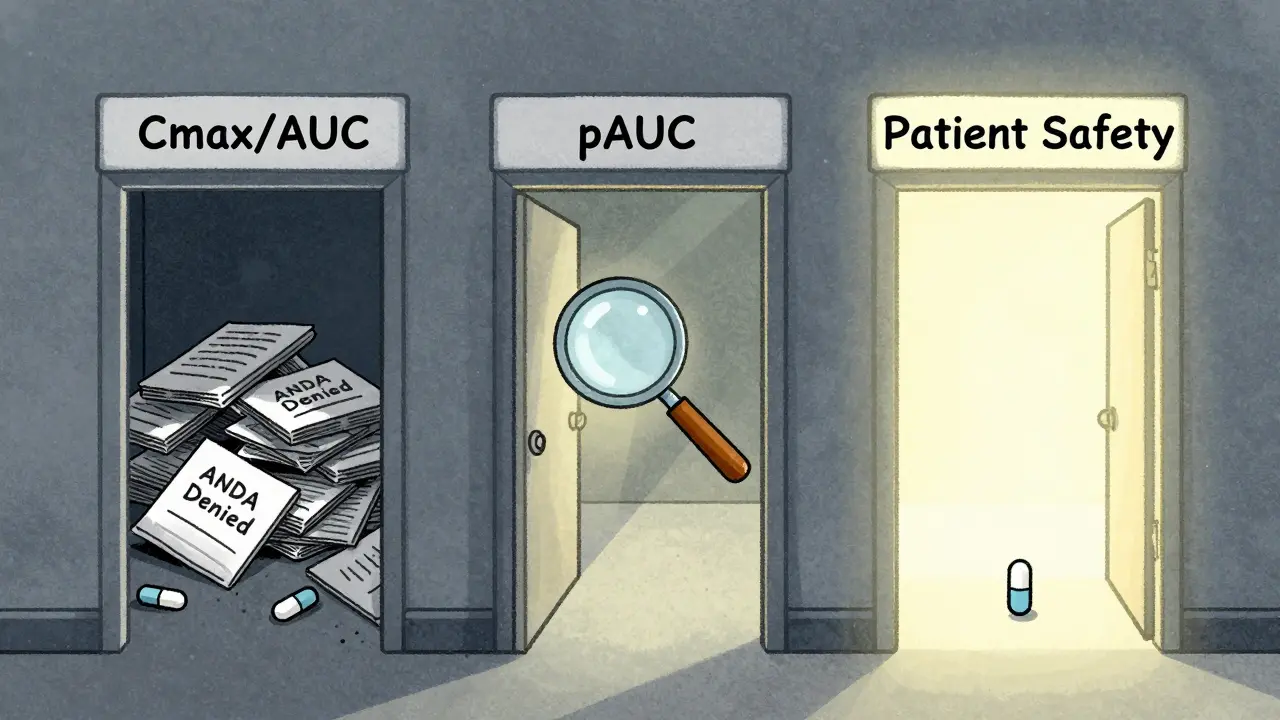
It’s not a quick fix. But for the right drugs, skipping pAUC could mean risking patient safety-or getting your ANDA rejected. In 2022, 17 ANDA submissions were turned down just because of wrong pAUC time intervals. That’s 8.5% of all bioequivalence-related deficiencies that year.
What is the main purpose of partial AUC in bioequivalence studies?
The main purpose of partial AUC (pAUC) is to measure drug exposure during a specific, clinically relevant time window-like the first 1-4 hours after dosing-where differences in absorption rate could affect safety or effectiveness. Unlike total AUC, which looks at the entire curve, pAUC focuses on the part of the pharmacokinetic profile that matters most for therapeutic outcomes, especially for extended-release or abuse-deterrent formulations.
How is the time window for partial AUC determined?
The time window for pAUC should be tied to a clinically relevant pharmacodynamic (PD) effect. Common methods include using the reference product’s Tmax, setting a fixed time like 0-2 hours, or using a percentage of Cmax (e.g., 0-50% of peak). The FDA recommends selecting the window based on when the drug’s effect begins or when risks like abuse or overdose are highest. Pilot studies and published PD data are often used to justify the choice.
Is partial AUC required for all generic drugs?
No, pAUC is not required for all generics. It’s only mandated for specific complex formulations where traditional Cmax and AUC metrics are insufficient. This includes extended-release opioids, abuse-deterrent products, mixed-mode drugs (like IR+ER combinations), and certain CNS or cardiovascular agents. As of 2023, over 127 drug products have FDA guidance requiring pAUC, and the list is growing.
Why is partial AUC more variable than total AUC?
pAUC focuses on a narrow part of the concentration-time curve, often during the absorption phase where individual differences in gastric emptying, metabolism, or formulation performance create more variability. Because it’s measuring a smaller segment, small fluctuations have a bigger impact on the result. This higher variability often requires larger study sizes-sometimes 25-40% more subjects-to maintain statistical power.
What happens if a generic drug fails the pAUC requirement?
If a generic drug fails pAUC, the FDA will reject the ANDA (Abbreviated New Drug Application). In 2022, 17 submissions were rejected specifically due to incorrect pAUC time interval selection or statistical analysis. This means the drug can’t be marketed until the sponsor redesigns the formulation or study, runs a new trial, and resubmits-adding months or even years to development time and increasing costs significantly.
Do regulatory agencies outside the U.S. use partial AUC?
Yes. The European Medicines Agency (EMA) was one of the first to formally recommend pAUC in its 2013 draft guideline for prolonged-release formulations. Since then, the EMA has expanded pAUC requirements to 27 specific product categories. Other agencies, including Health Canada and PMDA in Japan, are also adopting similar approaches, though implementation varies. Inconsistent standards across regions can delay global approvals by 12-18 months.






9 Comments
Henriette Barrows December 29, 2025
Okay but like… imagine being a patient on one of these extended-release pain meds and your generic suddenly hits you like a shot of espresso instead of a slow warm hug. Scary stuff. I didn’t even know this was a thing until I read this, but now it makes so much sense.
Russell Thomas December 31, 2025
So what you’re saying is the FDA is now playing whack-a-mole with drug companies by inventing new metrics every time someone finds a loophole? Classic. Next they’ll measure the emotional weight of the pill.
Alex Ronald December 31, 2025
This is actually one of the most important shifts in generic drug regulation in the last decade. I work in pharma stats and we’ve seen firsthand how pAUC catches failures traditional AUC completely misses. The higher variability? Yeah, it’s a pain. But it’s worth it. One client had a 40% early exposure spike in their generic opioid - total AUC looked perfect. If we hadn’t caught it with pAUC, people could’ve OD’d. This isn’t bureaucracy. It’s harm reduction.
Duncan Careless January 1, 2026
Interesting read. The inconsistency in time-window selection across FDA guidances is a real headache for global developers. We’ve had projects delayed because one region wants 0–2h, another wants 0–Tmax, and a third says ‘use pharmacodynamic data’ without defining what that means. It’s not just expensive - it’s chaotic. Harmonization is long overdue.
Samar Khan January 1, 2026
😭 this is why generics are dangerous. they don’t care about you. they just want to make money. the FDA is asleep at the wheel. 🤡
Amy Cannon January 1, 2026
As someone who’s spent the last three years analyzing pharmacokinetic profiles for CNS generics, I can tell you that partial AUC isn’t just a technicality - it’s a paradigm shift. The fact that we’re now required to justify time windows based on PD endpoints rather than arbitrary cutoffs means we’re finally aligning regulatory science with clinical reality. Sure, it adds cost, yes, it demands more sophisticated modeling, and yes, it forces smaller companies to partner with experts - but the alternative - approving products that could cause overdoses or therapeutic failure - is ethically indefensible. The FDA’s move toward machine learning-guided window selection is the right direction. We’re moving from ‘is the area under the curve similar?’ to ‘is the patient’s experience similar?’ That’s not just science. That’s patient-centered regulation.
Fabian Riewe January 3, 2026
Big fan of this post. I used to think all generics were interchangeable, but now I get why some are riskier than others. My dad’s on an abuse-deterrent opioid - I’m gonna print this out and show his pharmacist. Knowledge is power, y’all.
Jim Rice January 4, 2026
Wait - so you’re telling me a drug can have identical total exposure AND peak concentration… but still be unsafe? That’s not science. That’s magic. Who’s paying these statisticians? I demand a refund on my biology degree.
Nicole K. January 5, 2026
This is why you can’t trust big pharma. They just want to make you sick so they can sell you more pills. pAUC? Sounds like a scam to keep you on drugs longer.